2012年,CRISPR基因编辑横空出世。
当时,所有人都觉得这将会是生物科技领域最强大的工具之一,然而,在疗效与安全性问题的双重困扰下,CRISPR基因编辑exa-cel疗法在临床中运用步履维艰。
如今,经过了十多年的漫长远征,CRISPR基因编辑疗法终于站在了成功的边缘。日前,在针对CRISPR基因疗法召开的FDA专家会上,专家小组表示这种镰状细胞病疗法足够安全,可以用于临床。这也就意味着,这一疗法获FDA批准上市的概率相当大,最终答案将在12月8日揭晓。
看起来,2023年很有可能会成为基因编辑疗法的元年。
/ 01 / 最大的不确定性:安全性疑云
Exa-cel疗法是福泰制药与CRISPR共同研发的一款CRISPR基因编辑疗法,用于治疗严重镰状细胞病(SCD)和输血依赖性β-地中海贫血(TDT)。
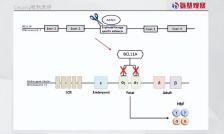
这一疗法的原理是,其可以利用基因编辑工具剪断骨髓干细胞中的一段DNA。这会释放被阻断的基因,以产生一种通常仅由胎儿产生的血红蛋白(HbF),而HbF指导正常血红蛋白的产生,从而治疗SCD和TDT疾病。
更重要的是,这种疗法仅需要输注一次即可起效,便能够解决严重镰状细胞病患者需要终身输血的问题。
在临床试验中,这一点也得到了证实。在17例接受exa-cel治疗并可评估疗效的严重镰刀型细胞贫血病患者中,94.1%的患者达到主要终点,100%的患者达到次要终点。
今年6月8日,FDA接受了exa-cel治疗SCD和TDT的生物制品许可申请。如果,FDA批准exa-cel上市,那么这款药物将会是全球首个批准上市CRISPR基因编辑疗法。
尽管exa-cel的疗效看起来没问题,但是FDA却对这一疗法的安全性问题有所疑虑,尤其是在“脱靶”效应上。
要知道,自2012年CRISPR基因编辑技术诞生以来,脱靶就成为其发展制约的主要因素之一。如果CRISPR基因编辑技术对预期靶标以外的其他DNA片段进行了切割,可能会破坏非靶向基因的功能或调节,引发严重后果。
为了确定福泰对这款药物脱靶的分析是否可靠,FDA召集了智囊团开会表决。
/ 02 / 新时代能否开启
10月31日,FDA召开外部专家会,以讨论该疗法的未来走向。
在会议上,专家小组表示,福泰对这款药物的脱靶分析相当详细,这种细胞病疗法足够安全,可以用于临床。
经过一番拉锯战后,基因编辑疗法exa-cel顺利通过了大考。
当然,这并不意味着exa-cel一定能够获批上市。虽然FDA最终是否会批准这款药物的上市,并不受到专家委员会的约束,但从过往的经验来看,大部分情况下,FDA都会遵循专家委员会的意见。
最终,exa-cel的终极命运将在12月8日揭晓。如果exa-cel能够顺利获批,作为全球首个获批的CRISPR基因编辑药物,对于整个基因疗法赛道的玩家来说,意义重大。在克服了安全性问题后,未来基因疗法将不断拓展治疗用途、打开更为广阔的想象空间,甚至带领整个生物制药进入新的天地。
不过,回到现实来说,exa-cel的获批上市只是个开始,后续的商业化会是更大的考验。
根据非营利组织临床和经济评论研究所的一份报告,如果该疗法获得批准,预计价格将极其昂贵,每位患者的费用可能高达200万美元。而高昂的治疗费用,很可能将患者阻挡在外。
这一点,蓝鸟生物上市基因疗法因未能与当地政府达成报销而从欧洲撤市,便是一个很好的例子。从这个角度来看,基因编辑疗法的未来仍然任重而道远。
但不管怎么说,CRISPR基因编辑疗法的成功都是值得期待的。毕竟,只有成功获批上市,踏出了第一步,才有资格去考虑未来的商业化。

原文标题 : 2023,CRISPR基因编辑疗法元年?