前言
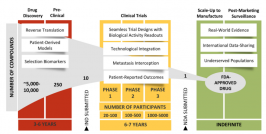
临床试验是发现和开发新疗法的关键引擎,它们是为最重要的问题提供客观和基于证据的答案的基石。在过去十年中,临床试验已经广泛发展,将癌症的生物学驱动因素转化为治疗机会。
当前,新一代临床试验的显著趋势包括从细胞毒性药物的评估转向分子靶向药物和肿瘤免疫药物的研究。从科学的角度来看,新药或药物组合的测试已经从经验主义转向假设驱动和基于生物标记物的研究。通过创新试验设计的应用和现代技术的整合,这些研究在患者选择和终点确定方面得到了加强。

制药公司在临床研究的所有阶段都发挥着越来越大的作用。监管机构通过在临床试验的许多方面提供指导,以及建立加速药物批准的新途径,对这些趋势作出了反应。患者报告的结果正在积极纳入临床试验,使用方便用户的仪器和数字工具。在很大程度上,临床试验中的这些变化是由于迫切需要为患者提供有效药物,同时保持对患者安全性和药物警惕性的密切监测。这些都需要所有利益相关者继续努力,以克服临床研究中持续存在的诸多挑战。
临床试验创新的科学考量
加强临床前和临床后的转化
一个药物的开发循环途径,包括从试验台到临床和临床后的反复反馈,可以在过程中的几个步骤中加快该过程。例如,体内和体外模型的分子特征可能揭示对试验药物或药物组合最敏感或耐药的组织学和基因组变化,从而提供对假定作用机制的额外见解。
美国国家癌症研究所(NCI)建立了一个患者衍生模型库,从治疗进展后的癌症患者中前瞻性收集的肿瘤活检和循环肿瘤细胞可用于创建患者衍生模型,以评估原发性和获得性耐药的机制。
分子、免疫和成像表征的现代技术
临床试验随着技术的进步而发展,技术进步提供了表征肿瘤细胞、其微环境和免疫环境的必要工具。使用二代测序(NGS)的肿瘤分子表征越来越多地在常规实践中进行,以实现生物标记物选择的临床试验。
尽管取得了这些进展,但试验合格标准仍然普遍狭窄,分子选择通常基于检测存档肿瘤标本中的单基因改变或蛋白表达;抗肿瘤疗效通常通过对靶病变的静态、线性测量来评估。未来,整合新的多维生物标记物,如全外显子组或基于基因组的突变特征,肿瘤免疫微环境中蛋白质或RNA的数字空间图谱,以及从标准护理成像中提取的定量特征的放射分析,将用于改善患者分层。
此外,人工智能(AI)和机器学习平台以及通过循环肿瘤(ct)DNA动态监测跟踪的能力可能允许通过自适应药物剂量测试个性化药物组合,从而平衡药物敏感克隆和耐药克隆之间的竞争性相互作用。
分子残留病灶的癌症拦截试验
在早期研究中确定安全性和耐受性后,通常对晚期转移性疾病患者进行新药物或联合用药的初步疗效评估。为了获得更大幅度的生存率提高,必须对接受明确治疗但复发风险高的可治愈恶性肿瘤患者进行有前景的方案测试。
癌症拦截是癌症早期的积极干预,提供了在临床复发前消除分子残留病灶(MRD)的机会。MRD描述了癌症衍生生物标记物可检测的状态,通常使用血液或其他体液中的高灵敏度和特异性分子分析,这些检测低于常规检测(如放射成像)的检测阈值。这种“防患于未然”的临床试验具有挑战性,这些研究不仅必须确定可从具有可接受治疗指数的额外干预中获益的患者亚群,而且通常需要长期随访,以观察基于时间的终点事件,如无复发生存率。
液体活检与超灵敏检测相结合以检测低水平ctDNA的出现导致了拦截临床试验的发展(例如ACTRN12615000381583、NCT03145961、NCT03832569、NCT04385368)。虽然并非所有有复发风险的肿瘤都会将ctDNA释放到血流或其他体液中,但量化这些释放的能力使ctDNA清除成为与无复发生存率相关的短期替代终点。将其他基于血液的生物标记物(例如基于血液的肿瘤突变负荷、免疫细胞比例)与ctDNA动力学相结合,可进一步提高免疫治疗反应预测的准确性。
另一个日益受到关注的领域是使用全基因组表观遗传学分析,同时评估液体活检中的多种癌症特异性DNA甲基化标记。在不同癌症类型和亚型的血浆ctDNA中可以测量到不同甲基化区域的不同模式。
临床试验创新的设计和方法考量
临床试验设计的取舍
临床试验分为几个阶段,以提供研究药物开发过程中继续或停止的关键决策点。至少有两个主要检查点用于决定是否应在大型随机III期试验中测试试验治疗。当安全性、耐受性和初步抗肿瘤活性得到确认后,第一个检查点是I-II期转换,以确定是否应在以组织学为基础或以分子为基础的中等规模队列(单臂或随机研究)中检查治疗的疗效。第二个检查点涉及主要基于完成重点II期试验时观察到的疗效信号的决定,该试验通常采用客观反应率(ORR)作为单臂研究的终点,或随机研究中的无进展生存率(PFS)。
尽管有这些检查点,但进入临床试验并获得监管部门批准的新抗癌药物的成功率依然很低。在过去十年中,出现了无缝的肿瘤临床试验,模糊了药物开发三个连续阶段之间的界限。在某种程度上,这种现象是由加快药物审批以转变癌症治疗的紧迫性所驱动的。如KEYNOTE-001、CHECKMATE-040的例子,这些试验在创纪录的时间内加速批准了有前途的抗癌药物。相反,在多个平行队列的大型、无缝I/II期试验中测试的许多其他药物未能产生明确的抗肿瘤活性,以告知未来的临床开发决策。
组织未定性的开发代表了近年来发展起来的另一个范例,这是因为人们越来越了解到,在多种肿瘤类型中,特定的致癌驱动因素或依赖性是共同的。组织学未定性篮子试验已使pembrolizumab在微卫星不稳定性高(MSI-H)或错配修复缺陷(dMMR)肿瘤或具有高肿瘤突变负荷的肿瘤中加速获得批准。
总之,临床试验设计不应是“一刀切”,剂量和时间发现研究的安全性和早期疗效信号的动态评估可能会告知下一步的最佳步骤。这可能会走特定试验阶段的传统模式,或演变为无缝或组织未定性的设计,以加快后续步骤。无论采用何种策略,研究人员都必须遵守既定的科学、道德和生物统计原则和标准,以确保数据完整性和研究对象保护。
适应性和敏捷性临床试验设计
医学创新的速度可能超过传统的随机临床试验(RCT),从而降低其结果的相关性。KEYLYNK-010(NCT03834519)研究就是一个例子,该试验在其生命周期内受到标准护理方案不断变化的影响。
该III期试验将接受雄激素信号靶向抑制剂和多西紫杉醇化疗的患者随机分为olaparib和pembrolizumab或对照组(abiraterone or enzalutamide)。在研究开始近一年后,2019年12月公布的CARD试验将cabazitaxel确立为新的护理标准,从而使KEYLYNK-010对照试验臂过时。
适应性研究设计,如多臂多阶段(MAMS)STAMPEDE试验(NCT00268476)中使用的设计或I-SPY2试验(NCT01042379)中使用的平台设计可提供解决这些问题的解决方案。MAMS设计允许针对RCT中的标准对照组同时测试多个感兴趣的药物(多组),根据适当的替代终点,在未显示足够活性的组中停止招募。相反,表现出足够活性的组可以继续招募,直到招募到足够的患者来评估主要终点。此外,使用MAMS设计,试验可以调整和添加新的感兴趣的疗法,而无需设计和启动新的单独研究。与多臂设计类似,多阶段(如II/III期)设计具有成本效益,因为它们可以灵活地将II期转化为III期,从而可以更快地获得结果,并且总体上需要更少的患者。
基于大数据和真实世界证据设计“智能”临床试验
随机对照试验是评价癌症新疗法的金标准。随着精准医学的兴起,RCT不可行的罕见适应症越来越多。对照组的缓慢累积或高退出率可能会阻碍随机研究治疗与主动对照组(标准治疗或安慰剂)的试验。
具有历史临床试验患者匹配数据或电子病历(EMR)的对照组可用于评估新癌症治疗的比较和成本效益。这些数据通常用于支持监管应用、标签扩展和公共资助医疗系统中报销的医疗技术应用。最近已经证明了聚合各种“大数据”来源以生成真实世界证据(RWE)以加速药物开发的价值。
美国食品和药物管理局(FDA)和加拿大卫生部最近发布了RWE在监管决策中的应用指南。RWE支持FDA批准的新适应症或标签扩展的成功例子包括用于费城染色体阴性B细胞急性淋巴细胞白血病的blinatumomab和用于转移性Merkel细胞癌的avelumab。
从RCT以外患者的经历中解读RWE具有挑战性。患者在癌症过程中经常在多家医院接受医疗护理,独立的EMR系统不相互连接以及很多报告需要手动整理以供研究使用。尽管存在许多复杂性,但已经成立了几个学术联盟,如癌症基因组图谱(TCGA)、美国临床肿瘤学会(ASCO)的CancerLinQ、美国癌症研究协会(AACR)的GENIE项目、莫菲特癌症中心的ORIEN和国际癌症基因组联盟(ICGC),以实现临床和基因组数据共享。
患者报告的结果(PROs)
将患者的观点纳入临床试验可以提供关于症状负荷、治疗相关副作用的耐受性以及干预措施对患者健康相关生活质量(HRQOL)的影响的重要信息。为了有效,此类患者报告的结果需要通过验证工具收集,如癌症治疗通用功能评估(FACT-G)和欧洲癌症研究与治疗组织生活质量问卷(EORTC-QLQ-C30),然后正确分析和报告。
然而,在临床试验中整合PRO存在一些挑战,例如,方案中PRO内容的描述和设计不足;延迟或少报PRO数据;数据缺失,尤其是由于疾病进展或药物相关不良事件导致身体不适无法提供PROs的患者;缺乏纵向数据收集,尤其是无法随访或无法亲自就诊的患者;以及评估从PRO项目实时收集的数据的临床可操作性。
临床研究中的新需求和优先事项
临床试验中存在的差距
在过去的几十年里,癌症研究已经超越了许多不同的挑战。主要优先事项始终是改善癌症的预防、诊断和治疗,使患者寿命更长,生活质量更好。然而,尽管随着时间的推移取得了重大的科学进步,但并非所有癌症患者都能平等地实现这些进步。一个关键领域是临床试验不平等,包括罕见肿瘤、弱势社会经济群体以及少数群体缺乏试验参与。罕见肿瘤占癌症诊断的20%以上,而临床研究投资比例极低。
癌症临床试验中另一个仍然存在重大差距的领域是定义什么构成了具有临床意义的影响。由于癌症研究成本高昂,衡量影响具有内在价值,投资回报只有在为患者带来具有临床意义的益处时才有用。欧洲医学肿瘤学会临床效益量表(ESMO-MCBS)是一种结构化的方法,它对抗癌治疗预期的临床意义效益的大小进行了相对排序。在包括694项随机临床试验的一项分析中,当通过ESMO-MCBS标准进行分析时,只有三分之一的报告良好的临床试验与临床有意义的临床益处相关。
临床试验的全球化
从历史上看,大型制药公司推动了低收入和中等收入国家参与的临床试验全球化,以最大限度地增加收益,降低运营成本,加快完成研究,支持新抗癌药物的开发和监管批准。
为了进行全球研究者发起的试验,研究设计可能需要考虑来自不同地理位置的项目的不平衡,以及种族、文化、生活方式、遗传特征、饮食和代谢的差异。入组标准可能因受调查患者群体在管辖区内可获得的治疗而有所不同。卫生条件、可用的支持性护理、地方性感染的存在以及患者群体的预期寿命会影响研究药物的提供和研究对象的安全性。最终,持续监督对于保证研究质量、保护受试者和正确解释临床结果至关重要。
提高临床试验的效率
为患者提供有效治疗的紧迫性要求设计灵活的临床试验,保证执行和科学效率,以解决最优先和影响最大的问题。在当今时代,启动、运行和结束临床试验的行政负担往往过大,需要许多监管程序。关键利益相关者(如研究人员、监管机构、赞助者和患者)的协作对简化临床试验流程至关重要,这对于加快肿瘤药物开发至关重要。
小结
进入下一个十年,临床试验参与的过程将是动态和适应性的,通过利用科学、技术和方法创新来预防治疗耐药性的出现。未来的临床试验将比以往任何时候都更加以患者为中心,因此必须整合患者的投入,以确保解决具有实际结果的最相关问题,从而提高癌症控制和治疗水平。下一代临床试验的成功将基于以下基本原则:基于全球的学习以促进实践,共同协作推进个体化治疗。
参考文献:
1.The Future of Clinical Trials Design inOncology. Cancer Discov. 2021 Apr; 11(4): 822–837.